JACC Heart Failure丨射血分数保留型心力衰竭(HFpEF)治疗新视角:从共病驱动的冠状微血管内皮炎症假说向脂肪因子假说的转变
射血分数保留型心力衰竭中共病驱动的冠状微血管内皮炎症假说向脂肪因子假说的转变
过去十年,解释射血分数保留型心力衰竭 (HFpEF) 的主流观点认为是由于多种合并症协同作用,触发全身炎症状态,导致冠状动脉微血管功能障碍、一氧化氮环磷酸鸟苷 (cGMP) 缺乏依赖性肌联蛋白异常,以及负荷依赖性心肌细胞炎症和纤维化。相比之下,最近提出的脂肪因子假说强调了一种合并症—内脏脂肪堆积来解释全身炎症、多种合并症和HFpEF的共存,并确定了一种特定的近端致病机制(即功能异常的脂肪分泌一系列促炎信号分子)。

研究表明,过多的内脏脂肪堆积和脂肪因子失衡不仅可导致 HFpEF,而且还可直接引起高血压、胰岛素抵抗、2型糖尿病和慢性肾脏病。内脏脂肪堆积和促炎性脂肪因子也能解释HFpEF的一些特征,而这些特征无法用冠状动脉微血管炎症假说来解释(例如,心房颤动、骨骼肌和肺部异常、肾脏钠潴留和血浆容量扩张)。脂肪因子失衡还会导致微血管功能障碍、cGMP信号传导缺陷以及肌联蛋白磷酸化异常,并且它们可以直接引起心脏肥大和纤维化。合并症驱动的微血管炎症假说未能找到一种选择性靶向心脏的血源性分子介质,相比之下,在脂肪组织中选择性地沉默特定的促炎性脂肪因子,却会对心脏产生远端效应,从而调节心脏结构并影响HFpEF的进展。
临床试验表明,增强一氧化氮/cGMP信号通路或具有非特异性抗炎作用的药物在HFpEF临床治疗中并未产生理想效果,而对HFpEF有益的药物(例如,胰高血糖素样肽-1受体激动剂、钠-葡萄糖协同转运蛋白2抑制剂)则能使脂肪因子分泌谱正常化。此外,尽管 60% 至 70% 的HFpEF患者存在冠状动脉微血管功能障碍(其中仅30%表现为内皮依赖性功能障碍),但超过 85% 至 95%的该疾病患者存在中心性肥胖(通过升高的腰围身高比评估)或内脏脂肪堆积(通过肠系膜脂肪或心外膜脂肪评估)。因此,与合并症驱动的冠状动脉微血管内皮炎症假说相比,脂肪因子假说提供了一个新的解释框架,具有更强的证据支持,并且适用于更广泛的HFpEF患者。当然也需要进一步的研究来支持这些观察结果。
01
关于HFpEF舒张功能障碍的早期误解
在早期,许多医生认为HFpEF是一种类似于遗传性肥厚型心肌病的疾病。肥厚型心肌病患者左心室舒张末期舒张压-容积关系被认为向上向左偏移,导致人们认为HFpEF中升高的左心室充盈压与“舒张功能障碍”有关。遗传性肥厚型心肌病通常见于年轻至中年男性,他们收缩压容易偏低,因为左心室无法充分充盈以支持每搏输出量。
然而,这种病理生理特征并不适用于老年HFpEF患者。老年HFpEF女性患者通常有高血压病史(而非低血压)。此外,尽管她们常表现出心室肥厚,但左心室腔大小通常轻度扩大(而非缩小),并且左心室舒张末期压力-容积关系通常不会发生偏移。事实上,随着HFpEF合并症数量的增加,左心室舒张末期压力-容积关系曲线逐渐向右移动,而不是向左移动 , 这表明左心室过度充盈,而不是指向被动心室顺应性受损(表1)。然而,尽管左心室舒张末期压力-容积关系曲线没有向左或向上移动,但多年来,HFpEF患者一直被描述为“舒张功能障碍”,HFpEF也被错误地称为“舒张性心力衰竭”。

表1:用于解释 HFpEF 中全身炎症和多器官合并症共存的概念框架定义要素和主要局限性
02
合并症驱动的冠状动脉微血管内皮炎症一氧化氮-环磷酸鸟苷假说在射血分数保留型心力衰竭中的发展
在20世纪90年代和21世纪初,医生们注意到HFpEF患者的两个有趣的临床特征。首先,患有HFpEF的老年女性通常除了高血压外,还伴有多种合并症。他们通常超重或肥胖,常有胰岛素抵抗或糖尿病,并且表现出许多非心脏器官功能受损,特别是肝脏、肾脏和肺脏。人们认为这些合并症可能起因果作用,因此治疗这些合并症可能缓解HFpEF。其次,HFpEF患者通常表现出显著的全身炎症,表现为C反应蛋白或其他循环炎症生物标志物的升高,组织层面则表现出促炎通路的上调,并伴有不同程度的间质纤维 化、冠状动脉微循环障碍和血管稀疏,这些已成为肥厚型心肌病(包括与高血压相关的肥厚型心肌病)的特征。
综合这些观察结果,Paulus和Tschöpe 于2013年将多种合并症、全身炎症和冠状动脉微血管功能障碍联系起来,并提出老年 HFpEF女性中出现的多种合并症——特别是肥胖、糖尿病、慢性阻塞性肺病、高脂血症和高血压—共同作用,触发了专门针对冠状动脉微血管内皮的全⾝炎症反应(图 1)。Paulus和Tschöpe提出,冠状动脉微血管内皮炎症可能降低邻近心肌细胞中cGMP和蛋白激酶G的生成,致心脏肥大和肌联蛋白磷酸化降低,从而引起心肌细胞僵硬度增加和心肌纤维化。合并症驱动的冠状动脉内皮炎症诱导的一氧化氮-cGMP缺乏可能是主要机制导致HFpEF的原因被称为Paulus-Tschöpe假说。该假说首次提出,HFpEF的致病机制不再是高血压,而是全身炎症。自该综述正式发表以来,关于该假说也有—些疑问出现。首先,尽管60%至70%的HFpEF患者表现出冠状动脉微血管功能障碍的证据,但射血分数降低型心力衰竭(HFrEF)患者也具有冠状动脉微血管功能障碍的特征 。 因此,这种微血管异常可能与左心室充盈压升高有关,而非左心室肥厚或全身炎症所致。其次,HFpEF中的冠状动脉微血管功能障碍通常与内皮无关。而且,在临床上几乎没有证据表明冠状动脉微血管内皮功能障碍与微循环内皮炎症相关。 HFpEF患者的心内膜心肌活检显示,心肌组织中存在轻度巨噬细胞浸润,促纤维化通路上调,以及能够触发炎症反应的内皮黏附分子表达增加。然而,这些变化是在未证实存在内皮炎症的情况下观察到的。在HFpEF患者的心肌组织中,显微镜下尚未发现冠状动脉微血管内皮炎症病变的组织学证据。第三,正如Paulus等人所认识到的,冠状动脉微血管内皮功能障碍并非实验性HFpEF中心肌细胞异常发展的先兆,因此可能并非其主要原因。第四,蛋白激酶G信号传导缺陷是实验性和临床HFpEF中报道的众多可逆细胞生物学异常之—,然而,健康心肌细胞中蛋白激酶G的表达通常也较低,且蛋白激酶G信号传导缺陷并非HFpEF特有。在HFrEF患者中也观察到蛋白激酶G信号传导异常。此外,肌联蛋白低磷酸化不仅见于实验性HFpEF,也见于实验性容量负荷过重状态和临床HFrEF中。因此,蛋白激酶G信号传导缺陷和肌联蛋白磷酸化在HFpEF中的临床意义尚不明确。这些异常常见于心室容量扩张(而非减低)的状态(如HFrEF)。第五,治疗干预的随机临床试验结果进一步揭示了一氧化氮-cGMP通路紊乱在HFpEF中相关性的重要问题(表1)。在NEAT-HFpEF试验(硝酸盐对射血分数保留的心力衰竭患者活动耐力的影响)和INDIE-HFpEF试验(无机亚硝酸盐输送改善射血分数保留的心力衰竭患者的运动能力)使用一氧化氮供体——单硝酸异山梨醇和吸入无机亚硝酸盐——治疗并未改善运动能力或最大摄氧量,且积极治疗伴随着通过加速度计评估的日常活动能力受损。
在VITALITY-HFpEF研究中(评估口服可溶性鸟苷酸环化酶刺激剂维利西呱改善射血分数保留型心力衰竭患者日常生活活动中的身体功能的疗效和安全性)和CAPACITY-HFpEF(可溶性鸟苷酸环化酶刺激剂普拉利西呱在射血分数保留的心力衰竭患者中的安全性和有效性)中,通过维利西呱和普拉利西呱刺激可溶性鸟苷酸环化酶实现的cGMP信号传导并未改善生活质量评分或导致临床改善。在RELAX试验(磷酸二酯酶-5抑制剂改善射血分数保留型心力衰竭患者的临床状态和运动能力)中,使用磷酸二酯酶-5抑制剂西地那非增加cGMP并未对运动能力和临床状态产生有益影响。无论—氧化氮和cGMP信号传导的药理学增强机制如何,临床试验结果表明,这种增强似乎对HFpEF没有益处,从而提示一氧化氮-cGMP信号传导不足不太可能是临床上HFpEF的驱动机制。
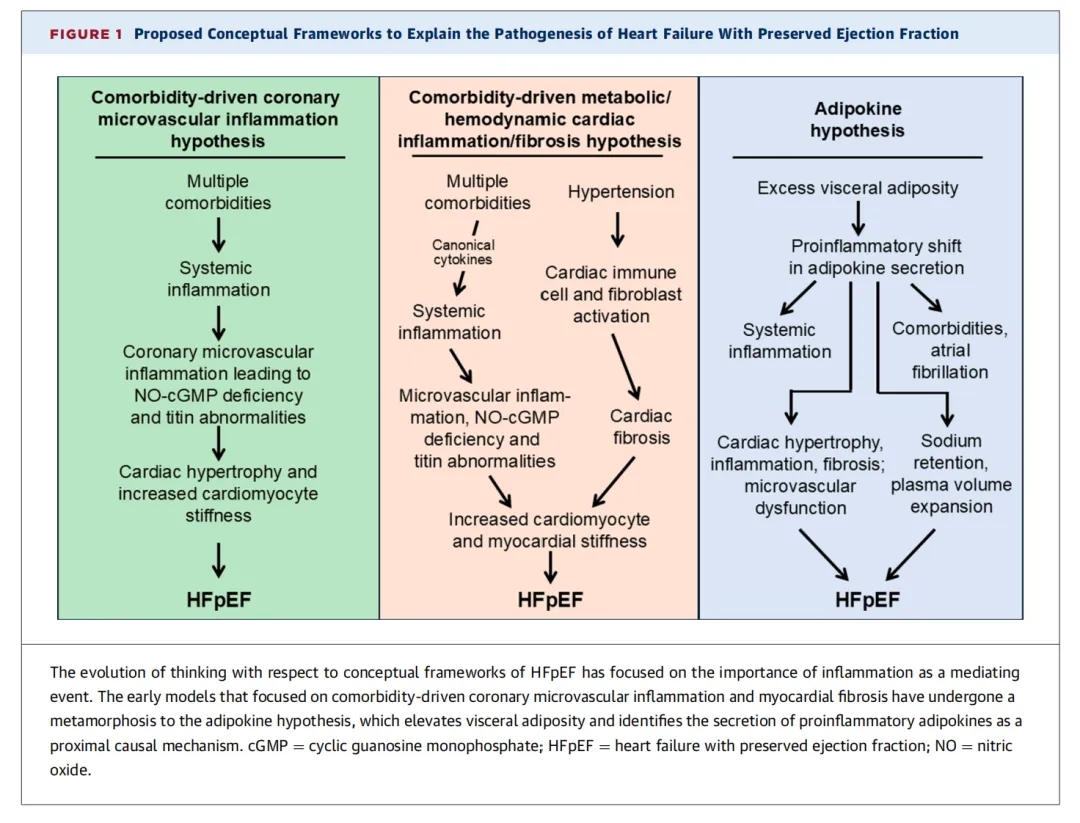
图1:HFpEF概念框架的思考演变
03
重塑Paulus-Tschöpe合并症驱动的代谢血流动力学炎症纤维化假说
针对这些担忧,Paulus-Tschöpe假说进行了重新调整,使其最初的关注点扩展到一氧化氮-cGMP信号传导和冠状动脉微血管炎症之外。一项修订后的框架(Paulus和Zile于2021年总结)提出,HFpEF的多种合并症(例如肥胖、糖尿病、慢性肾病和贫血)导致“代谢负荷”,从而促进全身炎症—这种炎症被认为是由经典细胞因子(例如TNF-α、IL-1和IL-6)和免疫球蛋白细胞黏附分子(例如ICAM-1和VCAM- 1)介导的(图1)。这些介质的作用可能导致冠状动脉内皮细胞中一氧化氮-cGMP耗竭和氧化应激,从而导致肌联蛋白磷酸化和剪接异常。而全身炎症(直接作用于心肌细胞)被认为会导致降解蛋白在冠状动脉内皮细胞中积累。此外,该框架提出,收缩期高血压会增加额外的“血流动力学负荷”,这可能导致成纤维细胞活化,并募集促炎和促纤维化的巨噬细胞和T细胞活化,从而导致细胞外基质蛋白依赖性的胶原蛋白在间质积聚。代谢负荷和血流动力学负荷的共同作用被认为会导致心肌细胞和心肌僵硬度增加。
然而,Paulus-Tschöpe假说的重塑—现在被描述为合并症驱动的代谢血流动力学炎症纤维化假说—并没有得到实验和临床试验结果的充分支持(表1)。 首先,归类于代谢负荷下的合并症(例如,糖尿病、慢性肾病和贫血)在生理上具有多样性,尚未被证实是促进全身炎症共同机制的上游病因,也未证实它们通过共同的细胞通路促进心肌细胞应激。 第二,在重新构建的框架中提出的HFpEF特定介质—生长分化因子-15水平升高—可抑制内皮炎症,并减轻HFpEF的心肌肥厚、心肌纤维化和舒张期充盈异常。这些有利作用与可能促进HFpEF发展的作用相反。 第三,多种合并症的存在并非HFpEF的独特特征,HFrEF患者也常见合并症,但尚未发现合并症在HFrEF中发挥特殊的病理生理作用。因此基于合并症聚类的表型映射尚未产生可重复的分组。第四,针对HFpEF合并症的治疗药物—例如降低2型糖尿病患者的血糖、改善肾小球功能或纠正贫血—并不能预测性地改善心力衰竭的临床进程,反而可能加剧病情。同样,减轻血流动力学负荷的药物(例如厄贝沙坦)似乎对HFpEF的临床进程没有产生有利影响,尤其是在当前时代。第五,针对经典细胞因子或细胞炎症反应的抗炎治疗尚未被证实对HFpEF有效。使用阿那白滞素(anakinra)拮抗IL-1并未改善HFpEF患者的功能,抑制IL-6也未能阻止实验性压力负荷过重或炎症诱导的心脏重塑,并未对心力衰竭产生有利影响。第六,秋水仙碱虽然能减轻全身炎症,但并不能改善已确诊心力衰竭患者的症状或预防心力衰竭事件的恶化。COLpEF研究的事后分析显示,使用卡那单抗选择性抑制IL-1β似乎可以减少心力衰竭事件,但这一观察结果与HFpEF的相关性尚不明确。第七,髓过氧化物酶抑制剂的抗炎作用(预期可最大限度地减少心肌细胞炎症反应)在随机安慰剂对照试验中并未对HFpEF患者产生有利影响。沙库巴曲缬沙坦抑制脑啡肽酶仅使循环胶原沉积生物标志物发生极其轻微的变化(尽管cGMP显著升高),且这些变化与药物对心力衰竭结局的影响程度无关。吡非尼酮的抗纤维化作用仅使心脏磁共振成像显示的细胞外液容量略有下降,且未对临床HFpEF患者的健康状况或舒张功能产生有利影响。
HFpEF 的脂肪组织-脂肪因子失衡假说
从2017年到2024年,一些研究者开始构建一个替代框架,以解释合并症、全身炎症和HFpEF之间的关联。研究发现,循环中瘦素水平升高和脂联素水平降低——这些激素通常仅由脂肪细胞分泌(即脂肪因子)—是实验性和临床HFpEF的共同特征。有趣的是,在HFpEF中观察到的瘦素和脂联素水平表达紊乱的特征模式,在患有各种合并症(例如高血压、胰岛素抵抗)的患者中也很常见,提示促炎和细胞保护性脂肪因子表达失衡可能是这些不同合并症、全身炎症以及HFpEF心脏结构和功能异常之间的媒介。 此外,一些研究者指出,这些脂肪因子的紊乱和HFpEF的进展似乎与心外膜脂肪组织和其他内脏脂肪库的扩张和生物学转化有关。因此,心外膜脂肪量的增加可能起到信号转导器的作用,作用于其相邻心肌的信号通路,促进促炎性脂肪因子的分泌,并将其作用集中于心肌细胞、心脏成纤维细胞和冠状动脉微循环。心外膜脂肪沉积也与心房肌病和心房颤动的发生密切相关。因此,心脏及其他器官周围的内脏脂肪库发生扩增,进而促使一系列促炎性脂肪因子分泌,这一机制似乎能够合理解释HFpEF的病理生理特征与临床特点。
脂肪因子的构建和测试
一项正式构建的脂肪因子驱动框架(于2025年提出)指出,肥胖和膳食营养过剩是HFpEF最相关实验模型的必要驱动因素,并且内脏脂肪和循环脂肪因子的变化在HFpEF发病数年前即可观察到,并能预测普通人群中HFpEF的诊断。重要的是,内脏脂肪的程度与HFpEF的严重程度相关。脂肪因子紊乱在中心性肥胖和HFpEF中同时发生,并与不良预后相关。根据脂肪因子假说,脂肪组织通过脂肪因子信号分子将其健康或紊乱的生物学状态传递给其他器官。2025年的框架极大地扩展了与HFpEF相关的脂肪因子数量,使其包含100多种信号分子,其中包括生物活性脂质和microRNA。实验性和临床HFpEF伴有脂肪因子抑制,这些脂肪因子可抑制心脏肥大、炎症、纤维化和微血管功能障碍,以及肾小管钠重吸收。相反,在实验性和临床HFpEF中,功能异常脂肪分泌的主要脂肪因子促进心脏肥大、炎症、纤维化和冠状动脉微血管功能障碍,同时导致肾脏钠潴留和血浆容量扩张,从而解释了与肥胖相关的HFpEF的特征性左心室过度充盈。脂肪因子假说与早期HFpEF合并症驱动的冠状动脉微血管心脏炎症模型有几个重要的区别。 首先,脂肪因子假说通过识别这三者共同的近端因果机制,解释了全身炎症、合并症和HFpEF中促炎基因的表达(既往被用来支持合并症驱动的理论框架)。其次,研究表明,过多的内脏脂肪和脂肪因子失衡可直接导致高血压、胰岛素抵抗和2型糖尿病、慢性肾脏病和心房颤动。有趣的是,脂肪因子失衡也会导致Paulus-Tschöpe假说中确定的所有病理生理异常。脂肪因子异常已被证实会导致微血管功能障碍以及cGMP和蛋白激酶G信号传导缺陷和肌联蛋白磷酸化异常。 此外,脂肪因子可以独立于对微血管的任何影响而促进心脏肥大和纤维化,这也解释了为什么冠状动脉微血管功能障碍并非心脏肥大和纤维化的必要条件。一些研究者在HFpEF合并症驱动的心脏炎症模型中发现的驱动心肌纤维化的机制—分泌型酸性富含半胱氨酸蛋白(SPARC)和组织金属蛋白酶抑制剂1(TIMP1),这些是已知的由功能异常的脂肪组织分泌的脂肪因子。HFpEF的这些附加特征并未在合并症驱动的冠状动脉微血管炎症假说中得到合理解释。 第四,脂肪因子假说确定了一系列特定的血液传播信号分子,这些分子在HFpEF患者中增加,并且在实验条件下已被证明会导致HFpEF。相比之下,Paulus-Tschöpe假说并未确定合并症单独或共同作用)可能直接驱动HFpEF发展的具体介质。第五,实验性HFpEF的特征是脂肪组织中促炎性脂肪因子选择性上调,这些脂肪因子仅在脂肪组织中产生远端效应,从而调节心脏结构和HFpEF的进展。相比之下,合并症驱动的心肌炎症假说并不依赖于HFpEF的组织选择性敲除和过表达实验模型。第六,胰高血糖素样受体-1激动剂(GLP-1RA)已知在HFpEF临床随机安慰剂对照试验中显示出有益效果,此外,钠-葡萄糖协同转运蛋白2抑制剂(SGLT2-i)和盐皮质激素受体拮抗剂(MRA)均能够直接作用于脂肪细胞,导致内脏脂肪不成比例地减少,同时还能使HFpEF患者特有的脂肪因子失衡恢复正常。这些发现与HFpEF临床试验中缺乏NO-cGMP信号传导缺陷的证据形成对比。第七,尽管60%至70%的HFpEF患者存在冠状动脉微血管功能障碍(其中仅30%的患者存在内皮依赖性功能障碍),但超过85%至95%的HFpEF患者存在中心性肥胖(通过腰臀比增加评估)或内脏脂肪堆积(如肠系膜、肾周或心外膜脂肪)。脂肪因子假说不仅适用于肥胖人群,更适用于内脏脂肪过多的患者,而内脏脂肪过多几乎是HFpEF患者的普遍特征。因此,与合并症假说相比,脂肪因子假说能够为更广泛的HFpEF患者提供解释框架。
HFpEF的脂肪因子假说标志着HFpEF研究的一个重要进展,它进一步证实了全身炎症是HFpEF发展过程中一个至关重要的机制。最初的研究框架侧重于冠状动脉微循环和NO-cGMP-PKG信号通路缺陷,但它但尚未明确具体的介导机制。相比之下,脂肪因子假说指出,内脏脂肪堆积是HFpEF的根本原因,并认为扩张和炎症状态的内脏脂肪组织分泌的促炎、促肥大和促纤维化脂肪因子不仅是全身炎症反应的分子介质,也是心脏肥大、纤维化和微循环功能障碍发展的分子介质。内脏脂肪堆积和促炎脂肪因子的分泌也被认为是导致HFpEF相关合并症(如房颤、钠潴留和骨骼肌异常)发生的致病机制。 与合并症驱动的NO-cGMP缺乏假说相比,脂肪因子假说得到了更多实验研究和随机临床试验的支持。
由于脂肪因子假说关注的是内脏脂肪过多而非冠状动脉微血管炎症,因此它可能也适用于更广泛的HFpEF患者。重要的是,脂肪因子假说确定了一系列新的靶分子,可用于开发HFpEF的新干预措施。
文献译自:10.1016/j.jchf.2025.102822
· END ·
专业的心血管医生学术交流平台


版权及免责声明:
本网站所发表内容知识产权归属医谱平台、主办方以及原作者等相关权利人,未经许可,禁止进行复制、传播、展示、镜像、转载、摘编等。经授权使用,须注明来源,否则将追究其法律责任。有关作品内容、版权和其他问题请与本网联系
发表留言
暂无留言
输入您的留言参与专家互动