Circ Res | 浙江大学梁平教授团队/复旦大学唐逸泉教授团队合作发现TMC6是病理性心肌肥厚的新型治疗靶点
病理性心肌肥厚通常由机械应力驱动,是导致心力衰竭的主要原因。但目前有效的治疗靶点仍十分有限。TMC (跨膜通道样蛋白)家族的研究主要集中于听觉、皮肤免疫及神经科学领域,其在心血管系统中的作用仍然未知。
2026年2月25日,浙江大学转化医学研究院/医学院附属第一医院梁平教授团队联合复旦大学脑科学研究院唐逸泉教授团队在Circulation Research杂志在线发表题为“TMC6 is a novel therapeutic target for pathogenic cardiac hypertrophy”的研究论文,首次揭示TMC6是病理性心肌肥厚的内源性抑制因子,提示增强TMC6活性有望为该疾病提供新的治疗策略。

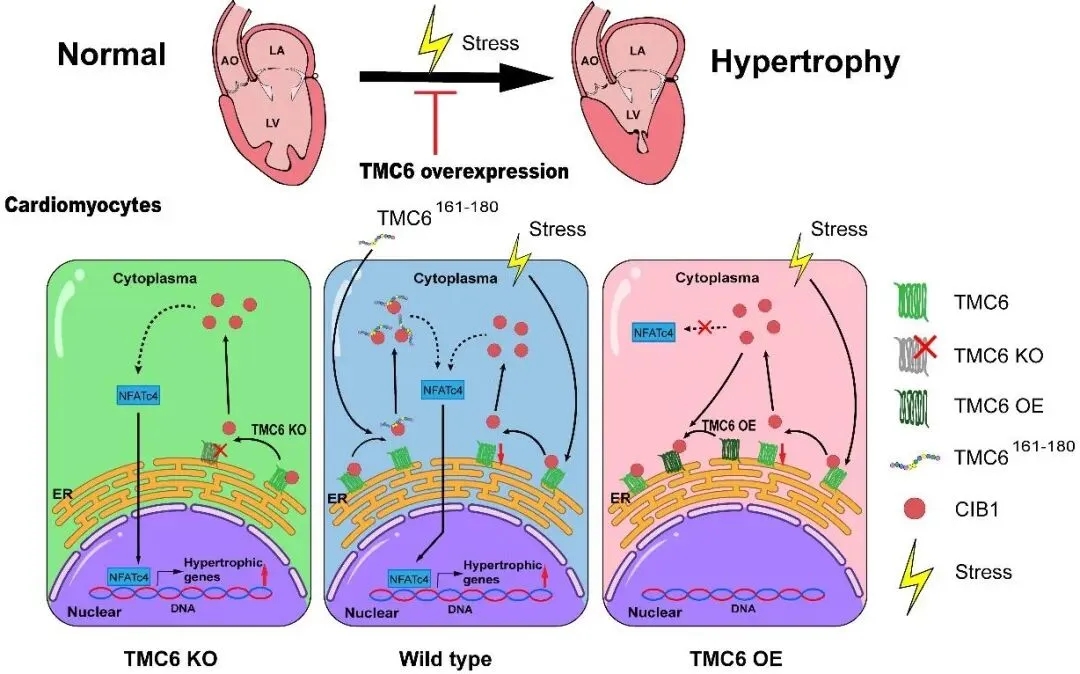
研究团队通过多维度实验体系展开系统探究,发现TMC6在正常心肌组织中表达丰富,而在肥厚心肌中表达显著下调,这一表达特征提示其与心肌肥厚的病理进程密切相关。为明确TMC6在心脏中的功能,研究团队构建了心肌细胞特异性Tmc6基因敲除小鼠模型。结果发现,TMC6缺失可诱发轻度心肌肥厚,并伴随心脏功能下降;在压力超负荷条件下,心肌肥厚程度及心功能减退进一步加剧。反之,过表达TMC6则可有效缓解压力超负荷诱导的心肌肥厚,改善心脏功能。
机制研究表明,TMC6定位于心肌细胞的内质网,其N端第161–180位氨基酸序列可与CIB1 (钙整合素结合蛋白1)发生特异性结合,从而将CIB1锚定于内质网中,进而抑制CIB1介导的calcineurin/NFAT信号通路激活,最终阻遏病理性心肌肥厚的发生。实验进一步证实,破坏TMC6与CIB1的结合可导致游离CIB1增加,从而启动促肥厚信号通路;而敲除CIB1则能够逆转因TMC6缺失所诱发的心肌肥厚表型,证实了CIB1是TMC6发挥抗肥厚作用的关键下游分子。
为评估TMC6的临床转化潜力,研究团队采用心肌特异性AAV9-cTnT-TMC6基因治疗策略,在压力超负荷诱导的小鼠心肌肥厚模型中进行干预。结果显示,恢复TMC6在心肌组织中的表达,能够有效重建其与CIB1的结合,抑制calcineurin/NFAT信号通路的活化,减轻心肌肥厚与心脏纤维化,并改善心脏功能。以上发现进一步支持TMC6作为治疗靶点的可行性与应用前景。
工作模式图
综上所述,本研究首次发现了TMC6在心血管系统中的全新功能,明确其为病理性心肌肥厚的内源性“刹车”分子;揭示了TMC6通过锚定CIB1,抑制calcineurin/NFAT信号通路,从而发挥抗心肌肥厚作用的新机制;同时证实了AAV9介导的TMC6基因治疗在病理性心肌肥厚中的潜在应用价值。这些发现既丰富了心肌肥厚的分子调控网络,又为靶向TMC6–CIB1轴的抗心肌肥厚治疗(包括药物与基因疗法)奠定了理论基础,有望为临床治疗病理性心肌肥厚和心力衰竭开辟新路径。
浙江大学转化医学研究院王洪坤博士(现为中国科学院杭州医学研究所博士后)、浙江大学转化医学研究院杨宗块博士和复旦大学脑科学研究院钟碧柔博士(现为科隆大学洪堡学者博士后)为本文共同第一作者。浙江大学转化医学研究院/浙江大学医学院附属第一医院梁平教授和复旦大学脑科学研究院唐逸泉教授为本文共同通讯作者。此外,本研究得到了浙江大学转化医学研究院陈静海教授、高峰博士、董晓璇博士,浙江大学医学院附属第二医院心内科马宏教授,以及复旦大学脑科学研究院唐逸泉教授团队王艳歌、范小治、张晓婷的支持和帮助。
原文链接:
https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.125.327680
· END ·
专业的心血管医生学术交流平台


版权及免责声明:
本网站所发表内容知识产权归属医谱平台、主办方以及原作者等相关权利人,未经许可,禁止进行复制、传播、展示、镜像、转载、摘编等。经授权使用,须注明来源,否则将追究其法律责任。有关作品内容、版权和其他问题请与本网联系
发表留言
暂无留言
输入您的留言参与专家互动